Programmable Medicine: Entire Cells as Therapies (The Century of Biology: Part II.II)
Cells, by nature, are extraordinarily programmable. They can be directed to differentiate from embryonic stem cells into any one of our body’s hundreds of cell types, to recognize and respond to various stimuli, to communicate with one another, among others. Scientists have made tremendous progress towards harnessing cells’ capacity to recognize and respond to stimuli to make living, programmable drugs to address complex diseases like previously untreatable cancers and autoimmune diseases.
Lying under the hood of this programmability is a complex network of circuits, across all scales from molecular to organ-level. It’s a system of intra- and inter-cellular communication, of checks and balances, of inputs and outputs. The below excerpt provides a great introduction to how the immune system’s circuitry works.
As an overview, our cells take a sample of all the proteins inside them, chew them up into little pieces, and display them on the outside of their cell membrane to broadcast its contents to our policing immune cells. These immune cells, trained on the viruses that are in and have been in the body, cross checks those protein pieces with their registry and kill any cells recognized as foreign. Doing so requires several regulatory steps:
- The T-Cell’s receptor (TCR) binds to the cell of interest’s antigen
- The TCR has two different binding sites, both of which must be turned on in order to activate the T-cell. This acts as an AND gate, increasing the odds it doesn’t identify a normal cell as foreign.
- If recognized as foreign, the TCR engages a signaling cascade, a positive feedback loop.
- That leads to the importing of the transcription factors necessary to express the genes coding for the activation, proliferation, and differentiation of T-cells from the nucleus.
- The activated T-cells release cytokines, a molecule that communicates to surrounding T-cells to join it on the front lines and attack these enemy cells.
- Various negative feedback loops are present at each step to act maintain balance and prevent an overreaction.
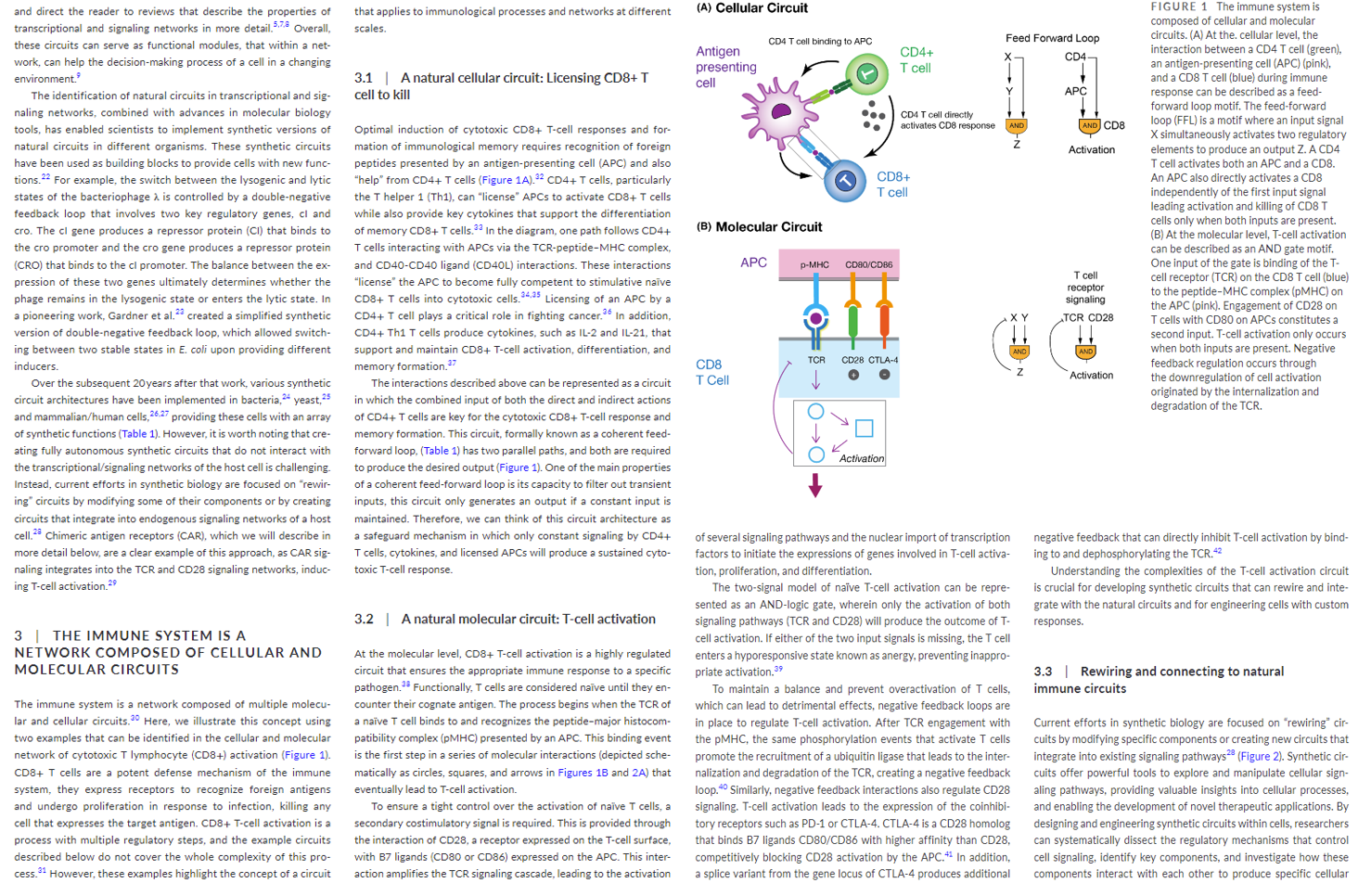
This intricate circuitry has been poured over by scientists to understand how to rebuild the machine to their desired specifications. This work has resulted in a Frankenstein-style T-cell that takes the receptor functionality from a B-cell, transplants it on a T-cell and programs this new cell called a CAR-T cell to target specific diseases like cancers. It works the following way:
Scientists hot-wired a patient’s T cells by removing them from their body, engineering them to express a novel Chimeric Antigen Receptor (CAR), and reintroducing the engineered cells to circulate throughout the blood detecting cancer cells. The CARs are a literal chimera of immune signaling components, with a portion of an antibody engineered to recognize cancer antigens strapped on to parts of the signaling domain of a typical T cell receptor.
The benefits of a living drug, in addition to being able to design complex circuitry, is that a single CAR-T cell can divide exponentially to kill hundreds if not thousands of target cells. Then, when the job is done and the foreign antigen is no longer detected, the army of CAR-T cells automatically contract to reserve status, patrolling for the target in the background for years.
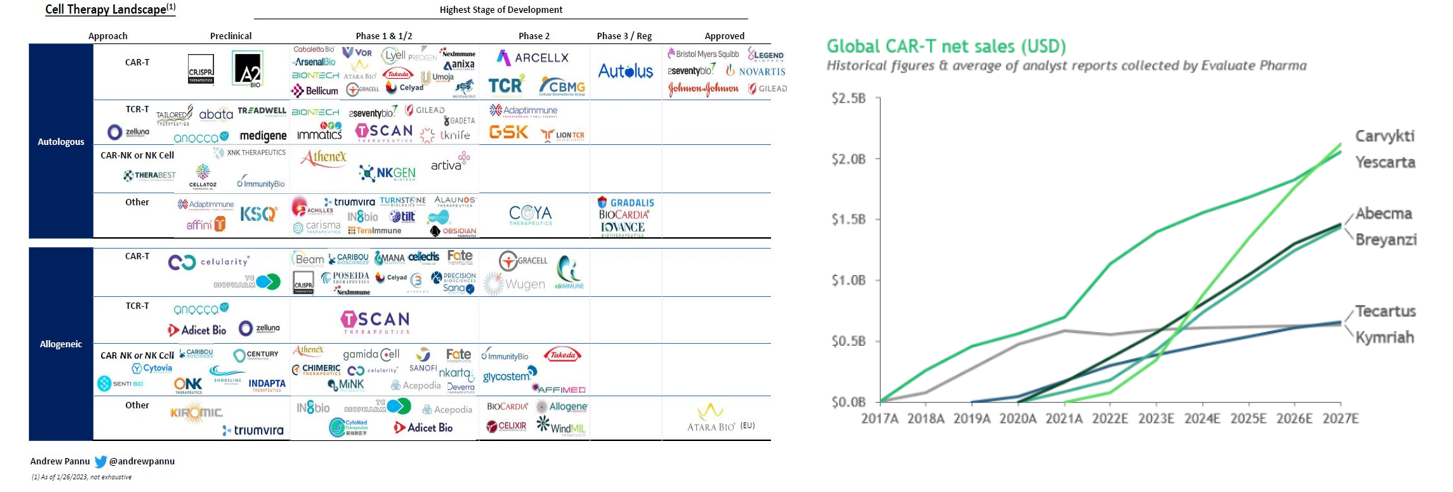
As seen below, the concept of hot wiring a T-cell has been iterated upon since 1989 and achieved its first clinical success in 2011 when it cured a leukemia patient, who remains cancer-free 12 years later. Now, the FDA has cleared six CAR-T cell products for 12 indications, across multiple antigen receptor designs. It’s had the most success combatting blood cancers, where the overall response rate across many trials in patients with refractory leukemia, lymphoma and myeloma is 50–90%. Moreover, while it has generally been used for advanced or relapsed cases, recent studies evaluating them in earlier lines of therapy have shown promise compared to standard high-dose chemotherapy. So far, 20,000 people have been treated with one of the approved CAR-T cells, and there’s been low treatment-related mortality and no reports of the cells transforming into malignant cells.
The number of approved drugs is only the tip of the iceberg. There are over 1,000 clinical trials registered, 500 trials currently underway, hundreds of companies developing such treatments, and many new approaches in the pipeline.
In contrast to CAR-T’s effectiveness at treating B cell malignancies, it’s had no clear successes in treating solid tumors, which comprise ∼90% of all cancer cases. Though some minor wins have emerged, these tumors remain far more difficult to treat. Solid tumor cells heterogeneously express antigens and much of the time the antigen is also found on healthy cells. The lack of specificity results in high on- and off-target toxicity. While killing of non-cancerous cells also happens with B cell malignancies, it’s far less dangerous because healthy B cells are expandable. Secondly, the environment enveloping the solid tumors suppresses the immune cells’ ability to communicate with one another, to survive, and to identify the tumor cells, among other things.
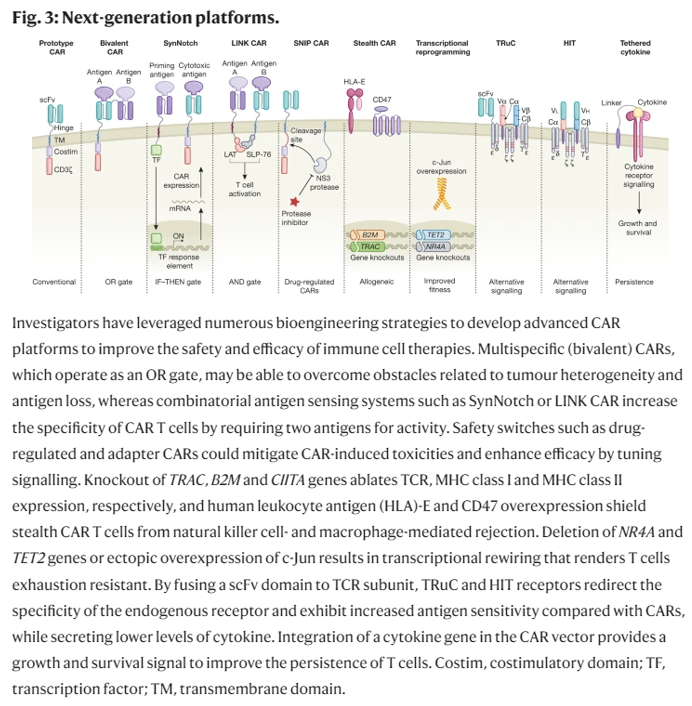
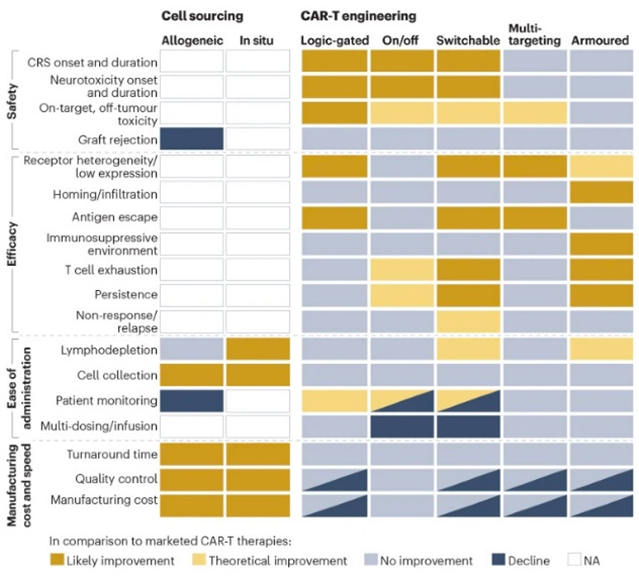
Researchers have devised new mechanisms to addresses these two challenges among others. These innovations aim to increase the CAR-T cells’ specificity, survival, or safety. They include logic gates like AND, OR, NOT; antigen density sensing; methods to overcome the tumor microenvironment; and, kill switches to trigger CAR-Ts to self-destruct on command.
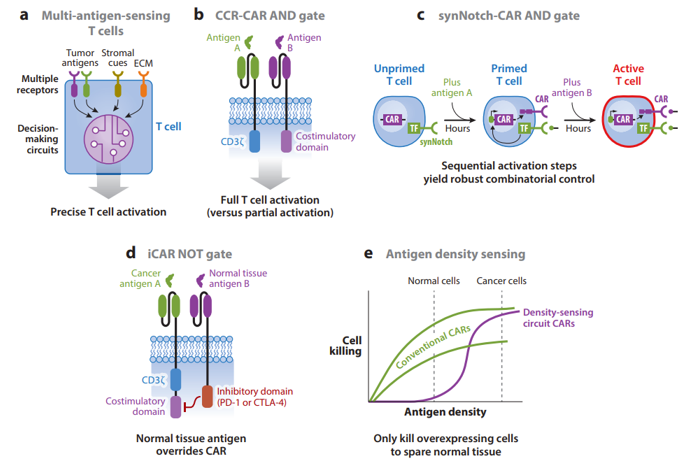
Pros and cons to each:
Specificity
AND gates started to be developed in the early 2010s. The most robust one built to date is synNotch, developed first in 2016. It works in the following way:
synNotch receptors (based on the native Notch receptor) use an extracellular recognition domain (e.g., scFv) to detect a target antigen. Binding the target triggers a proteolytic cleavage event that releases the intracellular domain of the receptor. In the synNotch receptor, this domain is a synthetic transcription factor that when released can enter the nucleus and drive expression of user-specified transgenes linked to the responsive promoter. Thus, synNotch circuits require transduction of the receptor and the response promoter. We have shown that T cells engineered with synNotch-driven CAR expression can function as highly precise and robust AND gates, sparing single-antigen but killing dual-antigen tumors in preclinical mouse models (Figure 2c). The synNotch-CAR AND gate strategy has shown significantly greater in vitro killing specificity for dual-antigen cancer cells than the CAR-CCR strategy because the synNotch-CAR mechanism of AND gating requires that the T cells transit through a series of sequential cell states (unprimed, primed, and activated), making activation by single-antigen cells very limited. The ability to precisely discriminate single-antigen cells could be very important when the CAR antigen is also expressed by a highly sensitive organ, such as the brain or lung. More recently, because of their flexibility and modularity, we have shown that synNotch receptors can be harnessed in combination with CARs and TCRs to generate a wide variety of different Boolean gates for diverse combinatorial antigen-sensing applications (W.A. Lim, unpublished data).
NOT gates have thus been underdeveloped with only one method being published so far:
It relies on expression of an inhibitory CAR (iCAR) that fuses an extracellular antigen-specific scFv to an intracellular inhibitory signaling domain [e.g., PD-1 (programmed cell death protein 1) or CTLA-4 (cytotoxic T lymphocyte–associated antigen 4)] (Figure 2d) (Fedorov et al. 2013). Coexpression of an iCAR with a CAR can prevent T cell killing of target cells that express the iCAR antigen. iCAR inhibitory effects are temporary and reversible, which is desirable for T cell NOT gates. However, iCARs’ ability to inhibit T cell killing is highly dependent on high receptor and antigen expression levels and the CAR signaling architecture. Thus, we need to develop more robust and versatile T cell NOT gates, as bioinformatic analysis of antigen expression patterns in a variety of cancers and healthy tissues suggests that negative regulation will be the most discriminatory component in combinatorial antigen-recognition circuits (O. Troyanskaya, personal communication).
OR gates to overcome tumor antigen loss or heterogeneity:
Another disadvantage of single-antigen-targeting CARs is that cancer cells within tumors often heterogeneously express antigens or downregulate target antigen expression during treatment, both of which can cause escape. For example, cancers can lose expression of CD19 via a variety of genetic resistance mechanisms, and a disappointing proportion of patients who initially respond to CD19 CAR T cells relapse due to antigen loss (Bagashev et al. 2018, Orlando et al. 2018, Sotillo et al. 2015). These relapses sparked the development of a variety of systems to program T cells with OR gates that kill based on recognition of either CD19 or CD22 (or another B cell antigen) (Zah et al. 2016, Zhao et al. 2019). Perhaps the most promising strategy is to engineer T cells with one CAR that has tandem antigen recognition domains separately targeting CD19 and CD22 (Fry et al. 2017, Schultz et al. 2018, Qin et al. 2018). OR gate CARs can also be built using a single binding domain that can bind multiple tumor antigens, which is usually achieved using a natural ligand as the targeting domain (Baumeister et al. 2018, Gilham & Maher 2017, Klampatsa et al. 2017, Lee et al. 2017). Other approaches include expressing up to three separate CARs (or a CAR and a TCR) targeting different antigens in the same T cell or dosing multiple T cells, each targeting a different antigen (Bielamowicz et al. 2017, Chen et al. 2017, Ruella et al. 2016, Slaney et al. 2017). However, expressing one CAR with tandem recognition domains has shown the best efficacy in preclinical mouse models (Hegde et al. 2016). Engineering T cells to overcome antigen heterogeneity is critical when targeting solid tumors. For example, in glioblastoma multiforme, a subset of patients express a truly tumor-specific antigen, deletion variant III of the epidermal growth factor receptor (EGFRvIII); however, EGFRvIII is heterogeneously expressed, and a CAR T cell clinical trial targeting EGFRvIII failed to observe any tumor regression despite evidence of killing EGFRvIII-positive cancer cells (O’Rourke et al. 2017). Thus, generating CAR T cell systems that are specific enough to prevent toxicities but also flexible enough to detect heterogeneous antigen expression patterns will be critical for treating solid tumors. We are currently exploring how synNotch-CAR circuits could be used to prime based on recognition of a heterogeneous antigen like EGFRvIII, but then kill based on a more homogeneous, though less specific, antigen. Overall, approaches that allow T cells to integrate information across multiple cells in a tumor would be powerful tools in overcoming heterogeneity, and could be another unique advantage of cell therapies.
Antigen density sensing to discriminate between cancerous and healthy cells:
Despite the safety of antibody therapy targeting ERBB2, a CAR built using this antibody, Herceptin® (trastuzumab), caused fatal toxicity due to targeting normal cells expressing low levels of antigen (Morgan et al. 2010). It is now known that CAR T cells can target antigens expressed at significantly lower levels than traditional mAb (monoclonal antibody) therapies, increasing the risk of toxicity (Stone et al. 2012,Walker et al. 2017,Watanabe et al. 2018). One approach to boost the specificity of cancer recognition is to incorporate antigen density sensing, as tumors often overexpress antigens found at lower levels in normal tissues. Density sensing was first demonstrated by lowering CAR scFv affinity to only recognize antigen-overexpressing cells (Caruso et al. 2015, Liu et al. 2015). An affinity-tuned T cell should robustly kill tumor cells overexpressing the antigen and spare normal cells expressing physiologic levels (Figure 2e). To achieve affinity-tuned CAR recognition of a wide pool of antigens, researchers will likely need to carry out specific screens to generate lower-affinity antibodies than are generated using conventional methods (Lim & June 2017). Our lab is exploring alternative methods to engineer antigen density sensing in T cells. New circuits that incorporate antibody affinity, receptor expression levels, and positive feedback loops can achieve even higher discrimination based on antigen density than by tuning CAR scFv affinity alone (W.A. Lim, unpublished data). Moving forward, combinatorial antigen recognition circuits could also be improved by integrating density-sensing capabilities.
Survival
The mechanisms above are meant to program the cells to only target cancerous cells. However, that’s only the first part of the journey towards effective therapeutics against solid tumors. Solid tumors dwell in living conditions that suppress t-cells and other cells coming to kill it. This dastardly tumor microenvironment (TME) can include physical barriers, heightened pressure resulting in compressive forces, lack of oxygen and nutrients, suppression of signaling capacity to communicate with fellow T-Cells, checkpoint inhibitors, among others. Researchers have been developing metaphoric armor, gas masks and snack packs for the T-Cells to survive the TME.
Locating the TME:
Solid cancers are outside of the lymphoid circulation; therefore, synthetically homing T cells to solid tumors could help. Making matters more difficult, tumors often secrete chemokines that prevent T cell homing (Harlin et al. 2009), and T cells often have incompatible chemokine receptors for chemokines found in tumors (Griffith et al. 2014). To enhance tumor homing, groups have cotransduced CAR T cells with chemokine receptors corresponding to chemokines found in particular tumors. This approach has been tested in preclinical mouse models of Hodgkin’s lymphoma, mesothelioma, and neuroblastoma (Craddock et al. 2010, Kershaw et al. 2002, Moon et al. 2011, Stasi et al. 2009). Rather than relying on endogenous chemokine/chemokine receptor pairs, our group has shown that fully synthetic systems can control cell motility. Using a G protein–coupled receptor modified to respond to a bioinert drug, we engineered immune cells with drug-directed migration (Park et al. 2014). This technology can be combined with a drug-releasing bead implanted at a disease site to direct only engineered cells to migrate toward the drug.
Breaking the physical barrier:
Solid tumors also have physical barriers that pose additional challenges to T cells. The ECM in solid tumors limits T cells’ penetration and aggregation (Peranzoni et al. 2013, Salmon et al. 2012). Heparan sulfate proteoglycan (HSPG) is an important tumor ECM component. By engineering CAR T cells to express an HSPG-degrading enzyme, a group achieved increased infiltration and antitumor activity in preclinical tumor models (Figure 3) (Caruana et al. 2015). However, without some form of local regulation, T cells with ECM-degrading payloads may cause toxicity.
Overcoming checkpoint and signaling inhibition:
One approach to address this hurdle is to reduce T cells’ inhibitory signaling capabilities. CAR T cells with PD-1 expression disrupted by Cas9-mediated gene editing (Rupp et al. 2017) or TALEN (transcription activator-like effector nuclease) editing (Menger et al. 2016) have enhanced the clearance of PD-L1-expressing tumors (Figure 3). Alternatively, CAR T cells engineered to constitutively secrete mAbs/scFvs blocking the PD-1 axis led to local accumulation of the immune checkpoint inhibitors within mouse xenograft tumors and enhanced clearance (Rafiq et al. 2018, Suarez et al. 2016). Another approach to overcome suppressive signals is to express dominant-negative receptors that act as ligand sinks and block signaling. TGF-β is a cytokine secreted by tumors that can cause apoptotic effects (Li et al. 2006); however, tumors can avoid the apoptotic effects by expressing a nonfunctional TGF-β receptor (Knaus et al. 1996, Park et al. 1994). Overexpressing a signaling-incompetent TGF-β receptor in CAR T cells has enhanced proliferation, cytokine secretion, and persistence in preclinical models (Figure 3) (Bollard et al. 2002, Kloss et al. 2018). Another approach to overcome suppression is to synthetically rewire a naturally inhibitory input to a stimulatory output. For example, expression of a chimeric molecule that swaps the cytoplasmic tail of the CD200R inhibitory receptor with that of the costimulatory receptor CD28 caused enhanced T cell proliferation and effector function in preclinical models (Kretz-Rommel et al. 2007, Oda et al. 2017). Other similar receptors that block inhibitory signals or transduce them into stimulatory signals have also been developed to help CAR T cells overcome TMEs (Figure 3) (Liu et al. 2016, Yamamoto et al. 2019). While these are promising ways to boost efficacy, interfering with these inhibitory checkpoint and cytokine pathways in engineered T cells will require tight regulation to avoid activating autoimmunity ( June et al. 2017).
Overcoming metabolic suppression: the high nutrient consumption rates of cancerous cells leaves slim pickings for normal ones to stay alive.
Modulating T-Cell metabolism has been shown to enhance function:
The glycolytic metabolite phosphoenolpyruvate (PEP) sustains NFAT (nuclear factor of activated T cells) signaling and effector function, and insufficient PEP levels cause diminished antitumor T cell responses. Overexpressing PEP carboxykinase 1, which catalyzes PEP production, in T cells led to greater effector function (Ho et al. 2015)
CAR costimulatory domain identity is plays a role:
For example, CD28 CAR costimulation drives T cell differentiation into effector memory cells with enhanced glycolysis, while 4-1BB costimulation promotes central memory T cell formation with increased respiratory capacity, increased fatty acid oxidation, and enhanced mitochondrial biogenesis (Kawalekar et al. 2016). CAR T cell studies have shown that CD28 signaling leads to early proliferation but decreased persistence, while 4-1BB signaling leads to increased persistence and lasting memory formation (Long et al. 2015, Zhang et al. 2007). There is a clear need for improved CAR costimulatory signaling to give CAR T cells the most metabolically advantageous system to balance proliferation and persistence.
Improving proliferation and persistence:
Inducing memory CAR T cells improve the balance between proliferation and persistence would enhance the therapeutic outcomes for all cancers. There’s a couple approaches so far:
- A new approach to solve this problem is to reduce the strength or duration of CAR signaling. One study showed that using CARs with a single immunoreceptor tyrosine-based activation motif (ITAM), rather than the three ITAMs normally found in CD3ζ, led to highly functional T cells with enhanced persistence in preclinical tumor models (Feucht et al. 2019). The reduced activation signal from the CAR with a single ITAM caused especially profound increases in efficacy relative to traditional CARs in experiments infusing very low T cell numbers.
- Another approach is to regulate T cells’ epigenetic profile to maintain a proliferative state long-term. One study reported that anti-CD19 CAR transgene insertion unexpectedly disrupted the epigenetic regulator TET2 gene in one T cell during the lentiviral vector–mediated integration (Fraietta et al. 2018). The TET2 gene disruption caused this single T cell to proliferate massively in the patient, and at the peak of response, 94% of CAR T cells were found to have expanded from that single clone. Demonstrating the ability to intentionally engineer this behavior, the group knocked down the TET2 gene and found that CAR T cell proliferation, persistence, and potency-enhancing effects were recapitulated. Similar effects on CAR function have been achieved by expressing altered c-Jun proteins in engineered T cells to drive optimal proliferative phenotypes (Lynn et al. 2019). While potentially promising, modifications that enhance T cell proliferation and persistence create the risk of causing T cells to become autoreactive or cancerous. Thus, it may be important in the future not to simply make constitutive changes to the T cells to increase their potency (i.e., constitutive expression or knockout of particular genes), but rather to make these changes inducible at the site of the tumor (Figure 3). Smarter T cells that can autonomously sense when and where to amplify their potency and durability may be ideal.
Safety
Strengthening T-cell survival is a great objective but could intuitively cause side effects if not controlled. While unceasing differentiation into ultimately cancerous cells is a concern, it should be placed within proper context. Only two instances of T-cell transformation out of many thousands of treated patients have been reported. The more common concerns are cytotoxicity and other side effects reviewed here. Since the earliest days of CAR-T development, scientists have been devising methods to turn the cells either off or on as desired once they’re in the patient. Boosting the therapy’s safety may be essential for its success on solid tumors because it could enable the ratcheting up of dosing levels when recent research has suggested that the dosing of T-cells for solid tumors may be grossly inadequate.[1]
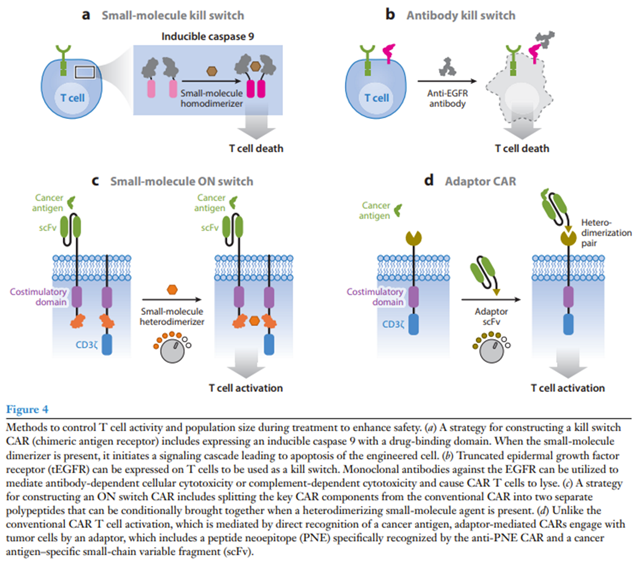
Drug-induced kill switch:
A popular kill switch for drug-induced CAR T cell apoptosis (iCasp9) was developed by fusing a modified human caspase 9 to the FK506 binding protein (FKBP) that dimerizes in the presence of a small-molecule drug (Straathof et al. 2005). An advantage of this system is that the drug eliminates only engineered T cells and ignores native immune cells, and it has shown efficacy in both preclinical and clinical contexts (Diaconu et al. 2017, Stasi et al. 2011). Furthermore, FDA-approved small molecules such as rapamycin can control iCasp9 switches with alternative heterodimerization domains and may face fewer regulatory hurdles (Stavrou et al. 2018).
Antibody kill switch:
Systems have also been developed that use antibodies to eliminate engineered T cells. Compact epitope tag constructs with low probability of natural immunogenicity were built for binding by FDA-approved antibodies. FDA-approved antibodies rituximab and cetuximab have been used with CAR T cells expressing the corresponding truncated antigen epitope constructs for both ex vivo cell selection and in vivo cell depletion and tracking (Figure 4b) (Philip et al. 2014, Valton et al. 2018, Wang et al. 2011). However, relying on antibody-dependent cellular toxicity to eliminate CAR T cells is risky in cancer patients with reduced immunity due to preconditioning or other previous chemotherapy.
On switch:
Rather than killing engineered T cells, another approach is to make CAR T cell activation dependent on a drug signal. Our group has developed ON switch CARs that need to sense both an antigen and a small molecule to activate T cells (Figure 4c) (Wu et al. 2015a). The ON switch CAR separates the key intracellular signaling components from the extracellular antigen binding domain on separate polypeptides that contain partner drug-inducible heterodimerization domains. The ON switch CAR is inactive until it senses both heterodimerizing drug and antigen. This system allows T cell activity to be titratably and reversibly inhibited.
Another group developed a system in which CD3ζ signaling is initiated by antigen binding while costimulatory signaling is independently controlled by a small-molecule dimerizer (Foster et al. 2017). T cells with this system are designed to fully activate only when both the target-specific CD3ζ signal and the small-molecule inducible costimulatory signal are received; however, like the CCR AND gate system, in vitro and some in vivo killing of CD3ζ CAR antigen–positive cells occur in the absence of the dimerizer drug. Still, the same group showed that this drug-dependent costimulatory system could be used in the same CAR T cells as an orthogonal drug-dependent inducible caspase-9-based safety switch (Duong et al. 2018). Robust, independent control of both CAR T cell expansion/persistence and death using orthogonal drug switches could enable strict modulation of therapeutic T cell population size throughout treatment.
Adaptors:
FDA-approved CAR T cells lack activity control after antigen binding and do not allow for retargeting of different antigens without using a new CAR T cell product. To address these limitations, researchers have split signaling and antigen-binding components between separate molecules in adaptor-mediated CARs (Cartellieri et al. 2016, Kudo et al. 2014, Ma et al. 2016). For example, in the peptide neoepitope (PNE) CAR system, T cells are engineered to express a CAR targeting a short, bio-orthogonal peptide tag. Under normal conditions, the PNE CAR does not bind target tumors (Figure 4d) (Rodgers et al. 2016, Viaud et al. 2018). When a tumor antigen–specific antibody fragment fused to the PNE tag is introduced, anti-PNE CAR T cells are redirected to kill the tumors cells. This system allows for reversible titration of CAR activity without the need to eliminate the CAR T cell. Furthermore, the same engineered T cells can change targets by adding a different tag-fused antibody.
Recently, a group developed a similar split, universal, and programmable (SUPRA) CAR (Cho et al. 2018). The SUPRA CAR system is a two-component receptor system composed of various pairs of universal orthogonal receptors expressed by T cells and corresponding tumor-targeting scFv adaptors that engage the receptors through leucine zipper interactions. Antigen targeting can be swapped by changing the adaptor, and signaling outcome is determined by the identity of the universal receptor. Like other adaptor-mediated CARs, the SUPRA CAR allows for reversible and titratable CAR activity, with the added ability to send combinations of signals to T cells for functions such as combinatorial antigen sensing.
See here for more detail on mechanisms to enhance safety
Additionally, using CRISPR-based gene editing strategies to engineer the cells can increase the manufacturing consistency[2] and to remove potentially toxic regions of the CAR-T cell:
The current standard approach engineers CAR T cells using viral vectors that permanently integrate transgenes in the genome (Esensten et al. 2016, Levine et al. 2017). Virally transduced CAR T cells experience random integration, making them less predictable and reliable. This may result in unintended consequences—such as clonal expansion, oncogenic transformation, and variable expression of the transgene—that could be avoided if the transgene were integrated into a specific locus. One study directed the CAR transgene into the TRAC (T cell receptor α constant) locus using CRISPR/Cas9 genome editing technology and found more uniform CAR expression with enhanced antitumor effects at lower doses of T cells, which could reduce side effects associated with infusing high T cell numbers (Eyquem et al. 2017).
Gene editing has also been used to inactivate toxicity-related genes in CAR T cells. Cytokine release syndrome (CRS) is a major CAR T cell toxicity (Brentjens et al. 2013, Brudno & Kochenderfer 2016). Although CRS can be managed with anti-IL-6Rα monoclonal antibodies or glucocorticoids (Le et al. 2018, Maude et al. 2014a), it can still be life threatening (Porter et al. 2018). To prevent CRS, a group inactivated the granulocyte macrophage colony-stimulating factor (GMCSF) gene, which has been known to promote CRS, in CAR T cells using TALENs (Sachdeva et al. 2019). Upon activation, engineered CAR T cells with inactivated GMCSF induced decreased macrophage-dependent secretion of CRS biomarkers without impairing T cell cytotoxicity or proliferation
A couple trials in the last two years have used gene edited T-cells, demonstrating their feasibility and safety, though not yet their potency or persistence. It will accelerate the research in developing new therapies, as numerous genes have been identified as possible avenues for enhancing T-cell fitness72,124,125,126,127,128,129,130,131,132,133.
As for clinical trials for the next-gen strategies detailed above more broadly, they’ve started in the most mature technologies: multi-targeting and armored constructs in blood cancers with autologous cells.
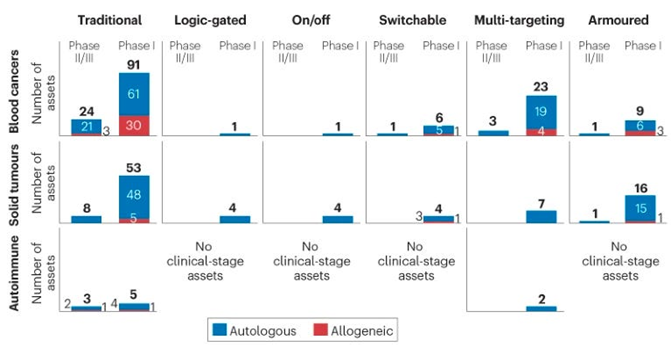
Here’s a list of all the clinical trials by category (i.e. each type of logic gate, fitness enhancements, engagement of the endogenous immune system, etc.)
One of the most exciting aspects about the future of cell therapies like CAR-Ts is that the techniques above are, to some extent, modular like Mr. Potatohead. They can be combined in one cell to address the specific intricacies of a given cancer’s tumor – for instance, an AND gate with a drug-induced kill switch and a fitness enhancement. For instance, one group combined a series of two synNotch receptors with a CAR to create a three-input AND and NAND gate. The quantity of mechanisms that already exist in this early phase of the technology’s lifecycle means that the combinatorial space of possible cells is massive.
Ongoing Limitations:
While CAR-Ts represent one of the most promising therapies in development, they have serious limitations at this stage, namely price. CAR-T therapies cost over $300K, with one approaching $1M to perform. These living therapies have the most complex manufacturing process ever devised, where T-cells must be isolated from a patient’s blood, genetically engineered, checked for purity, and then readmitted into the patient’s body. The excerpt below expands upon these manufacturing challenges[3]:
Autologous CAR T cells are personalized, living drugs that require a rethinking of the traditional manufacturing paradigm used for other biologics, such as recombinant proteins and vaccines111. The current approach to manufacturing autologous CAR T-cell therapies relies on a centralized model, in which patient materials are cryopreserved, shipped to the manufacturing facility, genetically modified and expanded, cryopreserved a second time, and then shipped back to the clinical site112,113. In addition to complex logistics, manufacturing of currently approved CAR T-cell products requires manual processing steps that must be conducted by highly trained personnel in good manufacturing practice (GMP)-certified facilities114. This cumbersome, centralized model presents a bottleneck for translation of preclinical candidates into phase 1 trials and will present challenges for commercialization of CAR T-cell and HSC therapies directed for indications with larger patient populations. It also presents challenges for translation in emerging markets that lack the required infrastructure for receipt and storage of cryopreserved materials.
In some of the first studies in which cell therapy was attempted, this manufacturing process took months and some patients even died during the waiting period. By the time the FDA approved the first CAR-T drug for broader use, the turnaround time had been reduced to 22 days. Despite these companies getting a minimal viable process down, the high prices remain due to drug developers needing to recoup their development costs (e.g. Novartis spent over $1B in R&D to develop the first approved drug), to the currently small scale of manufacturing plants, to the logistics of non-localized manufacturing, and to modifying the patient’s own T-cells rather than using more standardized cells. These issues are all being worked out. Big Pharma companies are investing heavily in infrastructure, as in the past year alone BMS invested in a 244,000 sq ft expansion of its cell manufacturing facilities and Novo Nordisk & Bayer invested a combined $386M in their facilities.1 These will help bring economies of scale to the centralized model. Meanwhile, startups are working on several futuristic solutions for localized manufacturing:
Going from Zero to One is the purview of startups, and a suite of new companies have emerged proposing futuristic solutions to the manufacturing bottleneck. Cellares, which recently closed a $255M Series C round and expanded their partnership with BMS, has built a new factory-in-a-box for cell-based therapy that they call the Cell Shuttle. Through a combination of hardware and software inventions, Cellares aspires to be an “Integrated Development and Manufacturing Organization (IDMO)” rather than a Contract Development and Manufacturing and Development Organization (CDMO) that meets demand by throwing existing technology and more humans at each new partnership. Whereas the new BMS facility required 500 new hires, technologies like the Cell Shuttle aim to produce the same volume of cell-based therapies with only 50. Carl June is hellbent on solving this problem—he’s an advisor and vocal proponent of Cellares, and has also co-founded his own stealthy CAR-T manufacturing company with $18M in backing from Danaher.
As if a network of robotic factories pumping out genetically engineered cellular medicines wasn’t a crazy enough idea, some startups are looking to bring manufacturing solutions even closer to the patient's bedside. A company called CellFE just raised a $22M Series A led by M Ventures (the venture arm of Merck KGaA) to continue to develop their microfluidic systems for cell manufacturing. The technology works by flowing cells along a tiny microfluidic chip where they are compressed as they flow through a narrow channel. When they re-expand, the molecular payload used to reprogram them flows into the newly available intracellular space. According to the founder, the vision is for cell-based therapy to evolve into a two-day process that is “manufactured and administered similar to what is done with dialysis today.”
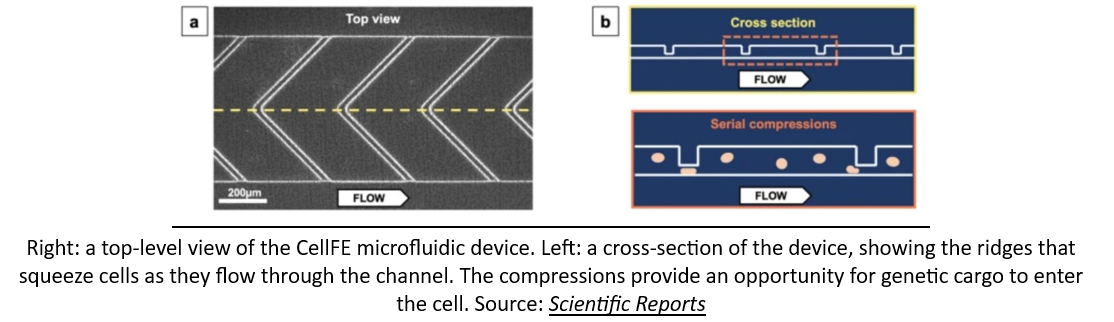
Additionally, groups are working on using standardized T-cells, other cell types, or even delivering mRNA that directs the body to make the proper immune cells for us (talk about local manufacturing!):
Several approaches are being pursued to address the manufacturing challenges for autologous CAR T-cell therapy. Off-the-shelf CAR T-cell products—whether derived from allogeneic T cells or iPSCs—have received considerable attention owing to the elimination of the complications associated with a personalized therapy, and are currently being evaluated in numerous clinical trials7,115. Results from one study this year demonstrated the approach to be feasible and safe in 43 patients with refractory multiple myeloma, with preliminary evidence of efficacy. Although the use of non-patient starting material considerably reduces the cost per dose and simplifies logistics116, manufacturing is still highly complex owing to the need for genetic editing117, and durability is not yet on par with autologous therapies7,118.
Several non-T immune cells, including natural killer cells, invariant natural killer T (iNKT) cells, γδ T cells and macrophages exhibit innate antitumour activity and do not induce GVHD, raising the prospect that they could provide an off-the-shelf source of cells with reduced toxicity, enhanced tumour trafficking and/or target antigen-negative variants through innate tumour recognition. However, allogeneic innate immune cells remain susceptible to rejection, raising concerns regarding the durability of their effects if they are not engineered for stealth. Cord blood-derived allogeneic natural killer cells incorporating ectopically expressed IL-15 have shown promise in a phase I trial for NHL and CLL138. iNKT-CAR cells mediated activity in mouse models, in part by cross-priming host CD8 cells towards tumour antigens195, and their safety and feasibility in a phase I trial for neuroblastoma have been demonstrated196. γδ T cells engineered to express a CD20-CAR have also shown impressive activity in early studies197. Expressing CARs in macrophages requires substantial adaptations of vectors and signalling domains, but antitumour effects associated with augmented phagocytosis, modification of the TME and recruitment of T cells198,199 have been demonstrated in preclinical models198, with CD3ζ-based CARs demonstrating equivalent phagocytic activity as Fcγ-based CARs
Efforts are also underway to create induced pluripotent stem (iPS) cell-derived CAR T cells200, natural killer cells201 and macrophages202. The differentiation of iPS cells to natural killer cells has been particularly successful, and clinical testing of these off-the-shelf therapies is currently in progress203, whereas iPS cell differentiation to fully functional T cells has been more challenging204. Given their nearly inexhaustible expansion potential, iPS cell-derived products could enable mass production of a homogenous cell product integrating numerous enhancements to endow stealth properties, safety switches and potency, and the long-term safety and efficacy results of these emerging platforms are thus eagerly anticipated.
Another method to mitigating the manufacturing bottleneck is the use of in vivo cell engineering, in which a patient’s T cells are transduced or transfected directly within the patient. In vivo cell transduction eliminates the ex vivo manufacturing step, and if successful, would enormously simplify the delivery of CAR T cell therapy and allow for translation to a broader patient population. To date, there have been multiple demonstrations of in vivo CAR T-cell generation in mice using CD4- and CD8-targeted lentivirus119,120, as well as an engineered adeno-associated virus121. Given the risks of off-target transduction, a potentially safer approach might be to transiently transfect T cells using T-cell-targeted lipid nanoparticles loaded with mRNA encoding the CAR. This approach was used to successfully treat cardiac fibrosis in mice using CD5-targeted lipid nanoparticles containing a mRNA encoding a FAP-directed CAR103.
Delivering modified CAR mRNA to T cells has been demonstrated by multiple groups. CAR mRNA was delivered to T cells through nanocarriers in mouse models of leukaemia and prostate cancer87. The mRNA-engineered CAR T cells performed similarly to retrovirally engineered CAR T cells by controlling the tumour and improving survival of affected individuals. Separately, it was shown that tLNPs could deliver FAPCAR mRNA to T cells in mice, resulting in CAR expression, reduced fibrosis and improved cardiac function in a model of heart failure, similar to the results of previous work with retrovirally transduced FAPCAR T cells54. Thus, a single injection of encapsulated RNA can produce functional and therapeutic levels of CAR T cells in vivo.
Applications in Diseases Beyond Cancer:
Though CAR-Ts have so far been optimized to treat cancers, their design principles and broad synthetic biology toolkit can and have been applied to an increasingly long list of diseases outside of cancer, including autoimmune, inflammatory, cardiac, infectious, and senescence-associated diseases. See these two studies for that full list, but the excerpt below give a sense for what’s possible:
Just cite this study that details possibilities of translation into among others.
- Autoimmune diseases
In contrast to cancer where the goal is to rev up the immune response, here it is just the opposite. The exemplar that this has promise is based on the recent publication of 5 patients with refractory lupus (systemic lupus erythematosus) treated with CAR-T directed against CD-19, documenting success of remission, that was maintained in approximately 1 year follow-up without concomitant drug therapy. Notably this was safe, as the major concern with CAR-T therapy are the cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity sndrome (ICANS), which can be life-threatening. Besides lupus, there are so many autoimmune conditions that can become refractory to medical therapies. Indeed, selective CAR-T intervention has also been recently applied to the experimental model of multiple sclerosis. This paves the way—it can be seen as just the beginning of selectively shutting down the untoward self-directed immune response in the future.
- Fighting fibrosis (scarring)
Last year there was a striking experiment in a mouse model of heart failure published in Science whereby the CAR-T cells directed against fibrosis were delivered via an mRNA-nanoparticle package. Remarkably, heart function was restored, as schematically shown below, with the scar-producing cells targeted by fibroblast-activation peptide (FAP) and shown to be cleared by the spleen.
Such fibrosis is a root cause of many fatal conditions that can affect most organs including the liver, kidney, lung, bone marrow and heart. They collectively account for more than 40% of deaths in the industrialized world. Again, while this is early work, it lays a foundation for a cell engineering approach to address a major unmet need. On a related front and perhaps more in the over-reaching category is the proposal to use CAR-T to target and reverse senescence as was suggested results from an experimental lung cancer model.
Moreover, CAR-Ts are just the beginning of engineered cell therapies. They are the proof-of-principle, the minimum viable product. What comes next? Well, for starters, there are lots of immune cells to be engineered. Many are already under active development as detailed in this review. Scientists may then begin to look beyond the immune system for opportunities. Once engineering cells becomes a standardized process, the next frontier may be designing systems of engineered cells that work together to cure more complex diseases. Cells would become modular in the way that the appendages of next-gen CAR-T cells like logic gates currently are. Eventually, these individual Mr. Potatohead cells will work together to form liquid organs, resembling a school of fish. Our bodies of course work in this way, with the prior sentence quite accurately describing the immune system as is. It divides up the labor amongst unique cell types and has them collaborate to address threats in a symbiotic way throughout the entire body.
Some groups are already working towards this goal, like the Hernandez-Lopez Lab at Stanford researching “biological circuits in multicellular systems and their applications in biotechnology.” The excerpt below details our latest work in this field.


The immune system is merely the first frontier of engineering cells to cure diseases. Parallel to the development of CAR-T therapies, decades of work has gone into the fields of stem cells and microbiome, and they’re both starting to show signs of therapeutic readiness:
Instead of testing new drugs in mice, why not test them in actual patient-derived cells? Some companies like Rarebase are already partnering with rare disease foundations to rapidly screen libraries of drugs in patient-derived iPSC models. Other companies like bit.bio are working to ship iPSC disease models for every cell type to researchers and companies as a product. They are also partnering with therapeutics companies to produce iPSC-derived cell therapies—including immune cells.
Much like for CAR-T therapy, researchers are asking the question: what if we could skip the cell isolation step and reprogram cells in vivo? This approach, called partial reprogramming, has emerged as one of the hottest potential longevity therapies. New longevity companies flush with cash like Retro Biosciences (backed by $180M from Sam Altman), NewLimit (backed by $40M of VC funding and $110M commitments from the founders Brian Armstrong and Blake Byers), and the goliath Altos Labs (launched with $3B in funding—the largest private biotech funding to-date—and a collection of biotech luminaries including Yamanaka) are all racing towards cell reprogramming therapies.
Ultimately, this won’t only be for cells, but will be the basis for entire organ systems—the full vision of regenerative medicine. Already, researchers in Israel have produced stem cell derived embryo models all the way up to 14 days of development. While this has immediate implications for the future of human reproduction—and companies like Conception are pursuing this application—it also opens the door to differentiating the embryos into other tissues (and potentially organs) with identical genetics to a patient in need of a transplant.
Alright… engineered immune cells to kill cancer, and iPSC/reprogramming therapies to cure, well… disease.
What about engineering the microbial cells and communities that inhabit the human body?
When you first learn that your own cells are outnumbered by your microbial inhabitants, it can be a bit disconcerting. In the most recent estimates, the human body consists of roughly 30 trillion cells—which is a lot, for sure—but not as many as the 38 trillion bacteria that live inside us. So far, the field of microbiome therapeutics has had a mixed track record. First met with extreme hype and a lot of funding, many of the first companies have experienced clinical setbacks. Trials have been placed on hold for safety concerns. Companies have died. Some of the early science has been called into question. Questions have been raised about the ability to produce consistent and reliable medicines, given that the first microbiome therapies have been sourced from human poop—a procedure called a fecal microbiota transplant. Seriously.
Despite the shaky start, there are signs that microbiome therapies are starting to reach a higher technology readiness level. To start, the FDA approved the first oral microbiome therapy in April earlier this year. Produced by the Cambridge-based Seres Therapeutics, the therapy (called Vowst) contains purified spores from a strain of bacteria capable of outcompeting another strain called C. difficile that can cause a serious gut infection. The fact that Vowst is going to be co-commercialized by Nestlé indicates that it is ready to be scaled. Name another company that ships top-notch espresso capsules, chocolates, and oral microbiome therapies. I’ll wait.
Closer to the frontier, things are getting even more sophisticated. It’s long been a goal to move beyond working with individual strains of bacteria. In the “third pillar” paper, the authors wrote: “In the future, the microbial community to be transplanted is likely to be an artificial, well-characterized mixture of strains.” Why? You don’t want uncharacterized poop, but using only a single bacterium is rate-limiting for complexity. A decade later, this is becoming a reality. In a massive effort from several top microbiome labs at Stanford, researchers created a stable synthetic community of transplantable microbes containing over 100 different organisms. Michael Fischbach, one of the “third pillar” authors, was a senior author on this new study. Talk about calling your shots. This type of synthetic microbiome will enable much deeper mechanistic science and more rationally engineered microbiome therapies.
A stable synthetic microbiome opens the door to some of the more futuristic ideas like engineered bugs that sense and respond to their environment, and even produce small molecules or biologics directly in the body. Given the mega-blockbuster sales of the first GLP-1 drugs (like semaglutide) for diabetes and obesity, this gets even more interesting. While semaglutide has been forecasted to potentially produce $300B in annual sales, the first generation of medicines has been shown to introduce the risk of severe stomach complications. For some of these medicines, up to 70% of patients experience serious side effects. A logic-gated microbial therapy that reduces stomach complications by tuning GLP-1 production in response to the gut environment could become a blockbuster cell therapy like nothing we’ve ever seen before.
This isn’t the only way that microbial and human cells could be used in combination. As if Dr. William Coley’s research wasn’t already vindicated enough, engineered microbes are now being used to boost CAR-T therapy. The mechanism is absurdly cool. Bioengineers from Columbia University engineered a strain of nonpathogenic E. coli to infiltrate tumors and deliver molecular “tags” that mark the cancer for detection and destruction by CAR-T cells.
If we zoom out to the limit, cell-based therapies are likely to be the endgame of modern medicine.
How can they not be? Every human starts out as a single cell. Through a profoundly complex cascade of genetically controlled cellular divisions, we ditch our microbial ancestors and grow into a mosaic of cells that are hierarchically organized into tissues and organs. Every disease stems from cellular dysfunction. By definition, if we can target—or even replace—diseased cells with other cells, it’s difficult to imagine a more effective medical paradigm. We’re talking about engineering a patient’s own immune cells to cure cancer, reprogramming cells and tissues to a younger and healthier state as we age, designing bacteria in our gut to pump out GLP-1 molecules when we eat too much, and curing diabetes with a single infusion of cells instead of countless infusions of insulin. If we continue to improve our capacity to program and produce cells, medicine will never look the same.
Read next section: pharmaceutical manufacturing scope and scale (addendum)
[1] Recent modeling of human and rodent circulatory systems has revealed that the delivery rate of CAR T cells to solid tumors is 10,000-fold greater in mice than in humans38, providing a potential explanation for the disappointing results of cell therapy trials in patients with solid tumors. This work challenges allometric-based approaches in preclinical studies that are traditionally used in dosing considerations for solid tumors39. While engineered T-cell therapy dosing has always required additional individualized features in the transition from mice to humans (for example, consideration of tumor mass and receptor expression), these findings suggest that improved dosing for solid tumors follows a less linear path than current algorithms suggest.
[2] ArsenalBio is one example of a company whose technology enables 1.5kb insertions into a specific part of the T-cell genome.