Neural Net Potentials: Near Frontier and Implications
While the entire world continues to look at transformers as the holy grail for all industries, we continue to believe that other novel computational approaches could prove as crucial in a variety of industries. This has led us to deeply observing Neural Network Potentials.
Modern NNPs are to molecular modeling as LLMs are to natural language processing. They’re flexible universal approximators based on GNNs or Transformers and learn the parameters over the course of training on ab initio simulation data.
They aim to be generalizable, near quantum level accuracy, and scale linearly with system size. In their 20 years of development, none has achieved all three simultaneously but they’re now getting close. They’ve already demonstrated density functional theory (DFT) level accuracy while being 5-6 orders of magnitude faster than regenerating that data from scratch and are starting to show meaningful generalization. Thus, they represent a major outwards shift in the Pareto Frontier of speed vs accuracy that limit current computational approaches.
DFT & CCSD(T) can generate near-quantum and quantum level accuracy, respectively, but are prohibitively expensive for anything above 100s of atoms and must be regenerated from scratch each time. Whereas, the same fixed functional form that makes classical FFs efficient means that they’re not transferable – i.e. they must be parameterized manually to each system being studied which requires months to years of expert time.
Lastly, the latest AI models are far faster but have a tenuous relationship to the underlying physics.
So, AI models can be thought of as tools for high throughput ideation or hypothesis generation and NNPs as useful for hypothesis validation, optimization, and mechanism elucidation.
The field of NNPs feels primed to reach an inflection point in the near to mid-term. The technical primitives are now largely in place and the models now have chemical accuracy with solid speed and thrilling glimmers of generality. Startups are being formed around the top models, Big Pharma & Big Tech are using them or developing their own, and the models are starting to show practical results.
David Baker’s lab used one as part of a high throughput virtual screen where it accurately predicted low energy state 85% of the time. Another group used a hybrid NNP/MM approach to estimate relative binding free energies to near chemical accuracy.
All of this, and scaling hasn't even reached the field yet. The models are still smaller than GPT-1. Some exciting models are even in the 10k’s parameter count.
Open Questions and their Implications for Company Building and Competitive Dynamics
Materials Science vs Drug Discovery
The first question to ask for company building is whether these general purpose systems are most useful for materials science or drug discovery.
In both industries, the true value is not in the technology by itself but the full stack integration of AI + NNP + lab designed to work systematically towards an end product. Orbital is a good example of a company not only seamlessly integrating computational excellence with a lab-in-the-loop but also understanding the limits of such automation.
Both industries also share the potential for individual asset sales or royalty-based, high-risk high-ROI joint ventures.
However, commercialization of innovative technology is a more well trodden path in biology; large, company-making deals are more common as opposed to reliance on many smaller ones; and, value capture via vertical integration is more attainable given that biology is closer to an information science whereas the value of materials science companies largely lies in Tesla's gigafactories or BASF's chemical plants – a capital intensity not readily stomached by investors.
Outside of the capital intensity required for value capture, there also are technical questions at play. The computational biology stack is more crowded with highly capable force fields developed over many decades. In comparison, the main computational approach used in materials science is running ab initio DFT, as classical physics can't model meaningful questions. That’s of course the ideal competition, because you have not a comparative advantage but an absolute one – NNPs are just as accurate and many orders of magnitude faster than the current alternative.
Moreover, meaningful materials science questions can be answered with small systems sizes – 100s-1,000s of atoms. Whereas, biological systems quickly become computationally intractable with even the fastest NNPs – e.g. proteins are millions of atoms.
To Scale or Not to Scale
There’s also a possibility that companies will pre-train their models on both materials science and drug discovery datasets if the models' returns to generality from scaling across elements and atomic environments is sufficiently strong. That of course doesn't mean that the company should open-endedly attack Li-ion batteries, industrial chemicals, and antibody drugs, but that universal models may be possible.
The models already show consistent scaling laws with training dataset size and training duration. And, the improved descriptors have laid the foundation for generality. Just as NLP models learned the fundamental latent structure of language, it’s intuitive that these models could go a long way to figuring out the lower level latent structures of interatomic interactions.
Indeed, what happens as these models scale up (which some teams are starting to do) may be the most exciting open question of those mentioned throughout this post. Will they display analogous power laws, showing ever improving quality and generality with scale? I.e., will the bitter lesson hold here too – and thus teams should focus more on scalability engineering than on descriptors, equivariance and other basic science? What are limitations of transfer learning – e.g. will a model’s training on heavy inorganic metals or radioactive elements help predict the dynamics of a protein folding? Will the loss in speed as they scale make them practically unusable?
The final question comes down to how long do the quality benefits from generalization (i.e. relative outperformance over specialized models trained on the given question of interest) outweigh the speed downsides. It’s a non-trivial question because while Microsoft’s 182M parameter MatterSim materially outperformed all specialized models with acceptable speed, a tiny model of 10Ks parameters went viral for its simulation of a crystal forming in water when that type of matter was entirely absent from the dataset.
It seems unlikely that company building dynamics seen in LLMs where a few labs suck in all the capital and talent like black holes will occur in no small part because the monetary reward comes so long after a sufficiently strong model is built. It took just two years for OpenAI to turn a good enough model (i.e. GPT-3) into a product pulling in billions in annual revenue. In comparison, the payback cycles in materials science and drug discovery even once you have the model are on the order of 5-15 years.
In those industries, it’s extraordinarily rare to have the privilege of vertically integrating from the get-go and thus capturing all the value you create (Moderna and CATL are relevant exceptions proving the rule). The more likely path is a slower build filled with joint ventures, partnerships or worse yet SaaS platform sales. This dynamic instills practical limits to the likely scaling these models receive.
Or, maybe we’ll wake up on a Tuesday to news that Google decided it wants to own the full computational stack of biology and materials science.
The counterargument to the scaling hypothesis is that small models will generalize sufficiently well to be better in practice than training massive universal models – which would probably have to be coarse grained for each given real world application anyway.
In such a world, NNPs’ end state would be similar to FFs today where there are several basic and reactive models with implementations for a bunch of different situations.
Will automation tooling advancements that make training small models quick and easy tip the balance towards this path? Can companies be built around small individual models? To what extent does the answer to that depend on the exact number of necessary models? For example, if you only need one reactive and one non-reactive organic model, then companies built on the best models could definitely thrive.
Such smaller models' smaller capital requirements would lower barriers to entry but also make the timeline to PMF quicker.
An intertwined question is how fast can these models get? Current optimized implementations of NNPs can achieve 2-10x improvement vs the base software, and that’s presumably from at most 1-2 years of work from a couple engineers. I also heard that the underlying MD software (OpenMM, LAMMPS, etc.) is antiquated and unoptimized, with speed ups achievable on the order of several times. In total, how far can the software be optimized? What about the hardware? Acellera’s transition to GPUs and optimization of software to them has accelerated their simulations by two orders of magnitude. Will an Anton be built for NPPs?
Data Needs for Scale
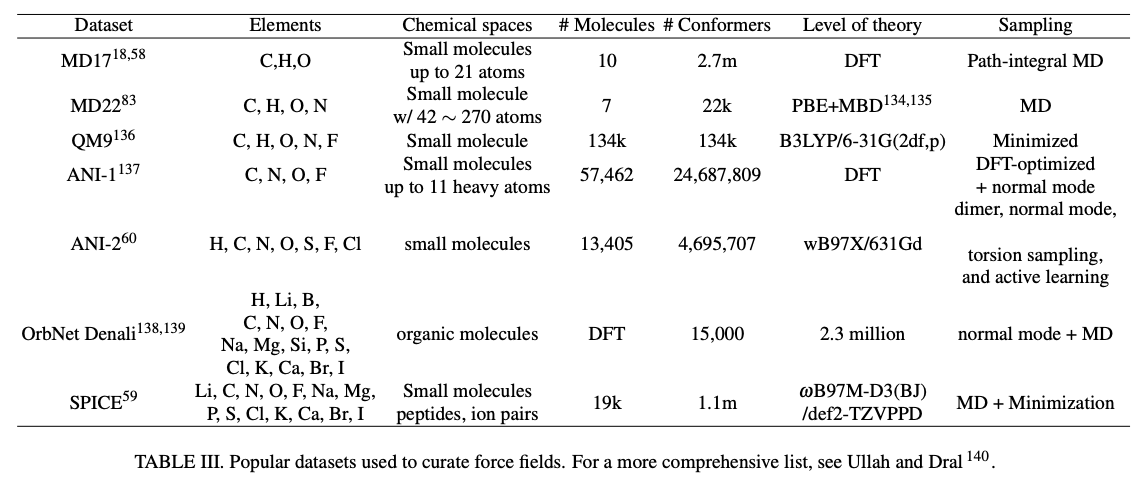
Just as we’ve seen with LLMs, another relevant unknown question with respect to scaling is how much data will be needed and how quickly can the datasets expand? While there are good resources (e.g. Materials Project, Open Quantum Database, SPICE 2, ANI-1x) for training and testing models, larger datasets with more diversity would be needed for orders of magnitude larger models. Researchers are hard at work trying to devise faster DFT algorithms and have made meaningful progress. Faster formulas are cheaper inputs to NNPs. Moreover, maybe it’ll be enough to train on data synthesized by another NNP and only generate new ab initio data when the models break down. Combining that strategy with further refinement on experimental data like free energies measurements should provide a reasonably cost effective way to scale data accurately.
NNPs' Role in the Computational Stack
NNPs are a symbiotic complement to AI models (e.g. AlphaFold3, ESM3, MatterGen), where AI models can be thought of as useful for high-throughput ideation and NNPs as virtual lab experiments. Or said differently, AI’s for prediction, while NNPs are for validation, mechanism elucidation, and to explore the most challenging areas of the conformational space that AI models cannot access.
AI models cannot explore these challenging areas due to a lack of data and a weak connection to the underlying physics. AI models can only predict based on their training data and there’s a seriously limited amount of data outside of equilibrium conditions. Hence, they won’t be a full-stack computational solution (at least unless they merge with NNPs) – i.e. they’ll long have their respective, separate purposes. While all this is true for both biology and materials science, I’ll explain what I mean with bio as the illustrative example as there’s been more published on the subject.
Models like AF3 are trained on the PBD, which is composed almost strictly of properly folded, crystallized proteins. This means they're at stable equilibrium / at minimum free energies and thus the forces on them are zero on average (otherwise they wouldn't be at equilibrium). Based on that fact, they assume that all the forces point back towards these equilibrium states where the forces are zero. Visually, they assume the PES looks like the left, though it may look closer to the right.

Hence, the models are effectively approximations of CG models – but only near equilibrium.
This makes them bad at approximating energies away from equilibrium. So they can't handle transitions, conformational switching no less dynamics or interactions through time, edge cases, protein folding, etc., etc. Moreover, their tenuous physics grounding means they struggle with situations largely absent from the PDB due to difficulty in crystallizing the structures – e.g. mutations, intrinsically disordered regions, orphan proteins, shallow MSAs, and novel sequences. They're not learning the physics to evaluate how two amino acids or two solvents will interact in a specific situation; they're saying based on the distribution of training data with two similar sequence motifs, this is the modal outcome of that distribution.
Unfortunately, AI models are already hitting a data wall. AF3 has consumed the entire PDB, a resource that took 50 years, $15B and many millions of PhD hours to make. There’s no next PDB. And, structure was a problem uniquely suited to AI in more ways than just the dataset.
With PDB being 50% of training data for AF3, the other 49.5% is a massive metagenomics sequence dataset. And while sequencing data is easier to scale, there are hints that sequencing data is seeing diminishing marginal returns. xTrimoPGLM pushed the sequence-only scale hypothesis, utilizing most metagenomic datasets that exist. It achieved SotA but was far from a step function. Basecamp Research is working on building a uniquely diverse dataset, but adding its 3M proprietary sequences still didn’t get us to the promised land. Moreover, the bitter lesson also hasn’t propelled small molecule models as they’ve scaled.
Another avenue is generating large-scale experimental data, like antibody screens or protein-activity datasets. AF3 included some creative things like adding high throughput experimental data for transcription factor-DNA interactions, but all those efforts still only represent 0.03% of training data.
There’s also the more fundamental question of whether these models are learning anything about the underlying PES as opposed to simply memorizing the motifs of interacting domains.
All of this is to say that even if it’s technically possible for AI models to glean all the physics knowledge necessary from structure, sequences and experiments alone (i.e. without training on any actual physics), it may be many many years before they have enough data to do so.
In the meantime, while I expect models to continue improving rapidly, I expect them to have a difficult time going beyond guesses to be validated and to long serve the purpose of virtual high-throughput ideation.
This leaves a large, valuable, and complimentary space for NNPs. They’ll examine with a fine-grained digital microscope the ideas that AI models generate and will be able to examine questions that will forever be impossible for AI models without a physics grounding.
Indeed, we’re already seeing signs of seamless integration of AI & physics-based methods. Researchers generated kinases in a range of conformational states using AF2, systematically explored those metastable states with MD and ranked the structures using Boltzmann weights, and then virtually screened the top structures with ligand docking. Other groups have seeded MD with AF-generated structures to explore cryptic pocket domain opening and ligand binding by or de novo generation of proteins in dynamic conformational states. Baker’s lab used AIMNet to identify low energy conformers and then a variety of AI techniques to generate macrocycles based on that.
These examples provide exciting glimpses of the type of company that we at Compound are excited by: a system that conducts high-throughput ideation with AI models, then uses NNPs to “zoom in” to explore the PES to validate the top leads, updating its hypothesis and plans based on what it finds on the PES.
How Those Open Questions Might Be Resolved Considering the Trajectory of the Field’s Frontier
All the questions above are ultimately about how the Pareto Frontier of generality vs accuracy vs speed may evolve and how that relates to company building and competitive dynamics – i.e. the intersection between technology and markets. Our job as investors is to not only understand such known unknowns but how they may be resolved and over what timescales. The field's frontier is rapidly expanding, both in terms of accuracy and speed.
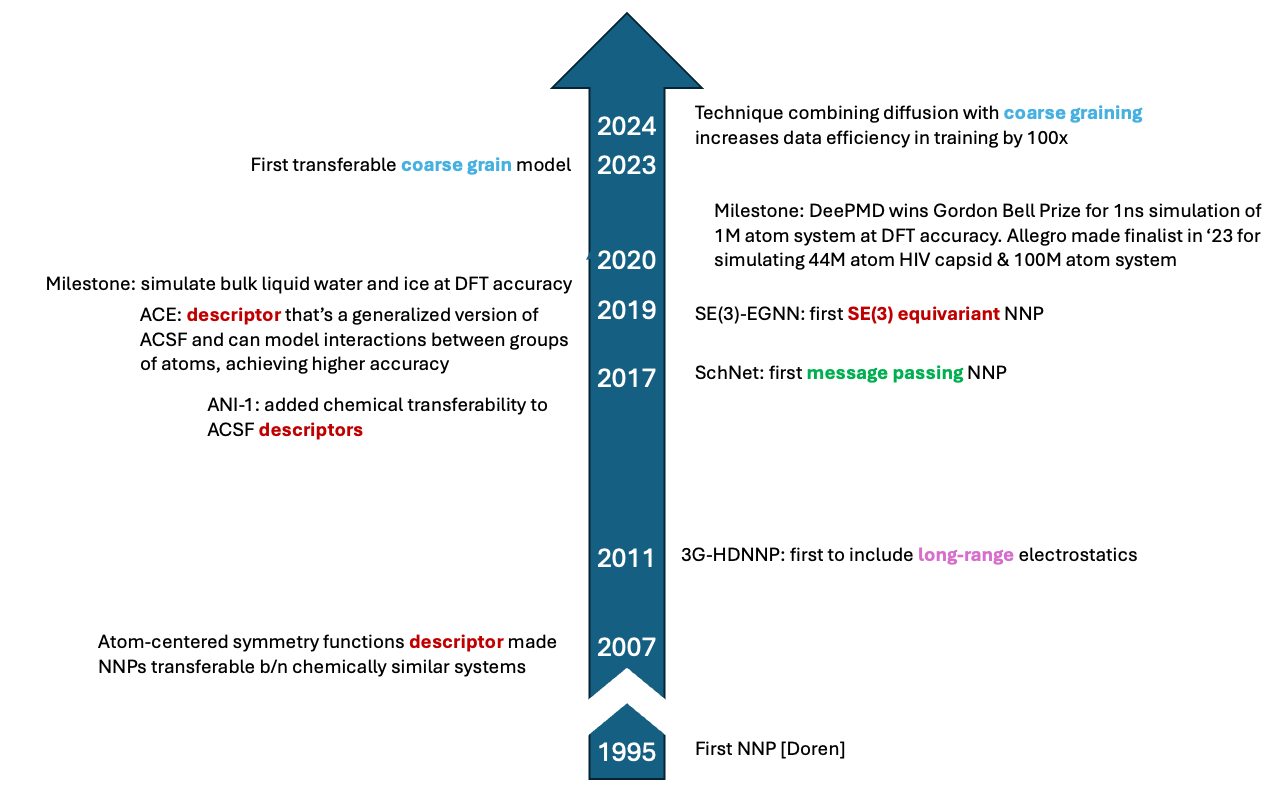
Accuracy and Generalizability
- Faithful Descriptors of the Atomic Environment
Estimating the energy of an arbitrary system requires faithfully transcribing the geometry of the 3D atomic environment into machine-understandable 0s & 1s and capturing all the relevant details about the systems – all without making the computation too slow. The former includes capturing the symmetries of space (e.g. when you rotate atoms, the energy doesn’t charge), the relationships between atoms (e.g. the angles between them), spatial patterns (e.g. rings, heteroatomic bonds). The latter includes whether each atom is charged, polarity, and other chemical properties. How generally yet specifically can you represent the atomic environment?

The first breakthrough came in 2007 with Behler and Parrinello’s atom-centered symmetry functions, which treat each atom as the center of its universe, encoding its local environment in a way that’s symmetrically invariant. The total energy is then estimated as the sum of individual atoms’ contributions. This enabled the first transferable NNP to similar systems, but the descriptor set was still hand crafted for each pair of elements. Advancements building on ACSFs have added the capacity to include chemical detail to make it increasingly generalizable across the periodic table and chemical systems. Atomic cluster expansion further generalized the descriptor by modeling interactions between groups of atoms for more accuracy. Other descriptor advancements include incorporating spin-multiplicity, polarity, etc.
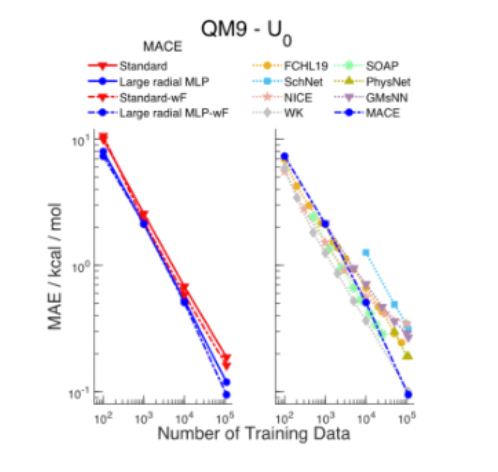
Each step function advance in our ability to mathematically represent the atomic environment with fidelity increases the models’ accuracy, generalizability, training efficiency, and stability. For instance, recent models that include SE(3) equivariance require 100 - 1,000x less training data to reach the same accuracy – crucial given the scarcity of publicly available training data in this space.
The price paid for such generality is speed. Thus, the cutting edge work in descriptor sets is to make descriptors more computationally efficient. Researchers have developed frameworks to operate in Cartesian coordinates not spherical ones which saves significant memory bandwidth and other groups are working towards more efficient representations of equivariance.
The steelman counterargument to all the above is that synthetic data from NNPs combined with fine tuning on DFT & CCSD(T) will relieve the severe data scarcity and from there the bitter lesson will hold: maximally universal and scalable architectures like graph Transformers will beat models with more fancy descriptors and equivariance.
- Message Passing Networks
Invented in 2017, message passing approaches learn the descriptors themselves or simply their parameters directly from the data. They treat molecules as 3D graphs, where atoms are nodes and bonds are edges. Employing end-to-end learning, they iteratively process info packets from neighboring atoms through the network and store the information in a vector or tensor.
One thorny issue is defining how many neighbors to communicate with. One must choose an arbitrary cut off, and as seen below, expanding the radius increases the number of neighbors exponentially. MACE and Allegro choose to only pass messages within the innermost circle. Remaining strictly local enabled GPU parallelization and thus scaling to entire supercomputers (because you can fit a full atomic environment on one GPU) but restricts ability to model long-range interactions. Not compromising in this regard is a core scientific frontier.
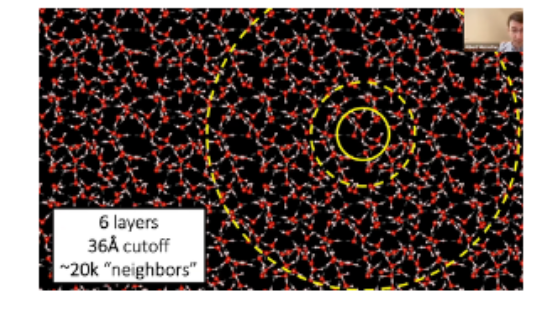
- Incorporating Long-Range Interactions
Efficiently incorporating long-range interactions in general is a challenge on the frontiers of science. These interactions include charge transfer, polarization, and electrostatic or dispersion. They’re necessary when modeling the most challenging PES like large protein structures, heterogeneous bulk or condensed phases, hydrogen bonding, excited states, etc. Hence, they’re relevant for commercial activities like protein folding dynamics, small molecule bonding, or most meaningful materials science problems. While researchers have worked towards incorporating these efficiently (e.g. electrostatics with quasi-linear scaling), the challenges remaining may be among the most difficult. It will probably never be completely solved such that the end state will look like FFs where there are separate reactive models that include long-range interactions.
- Training Approaches (e.g. Active Learning)
The last major advance in accuracy and generality came from approaches to training. Active learning automatically generates more of the data in areaş the model struggles with, thus systematically creating more diverse and high quality datasets. As parts of the potential are trained, the system estimates its own uncertainty and automatically generates new ab initio training data where it fails.
Another recent paper took the concept of “fine tuning” on CCSD data when necessary a step further by planning the entire training process as iteratively increasing the level of specificity. They first train on low-level “latent space” from a variety of lower tier, highly efficient data. Under such a transfer learning approach, NNPs can achieve better than DFT level accuracy. A final exciting recent approach is training a coarse grained (CG) model using diffusion, which boosted data efficiency in training by 100x.

All together, the models have achieved chemical accuracy with increasing signs of generality across chemical elements, size scales, temperature, phases of matter from gas to liquid, and types of structures from crystalline to amorphous.
Computational Efficiency and Speed
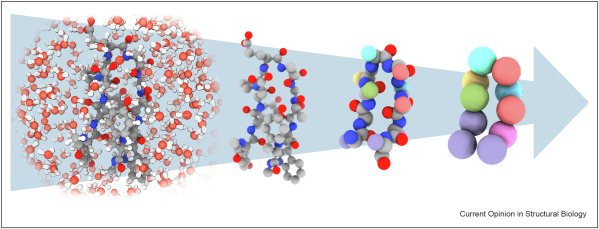
A variety of approaches seek to do the minimal computation necessary, modeling only the most crucial parts of the system with full accuracy and abstracting away everything else. These include coarse graining models, implicit solvent models, hybrid NNP / FF, hybrid diffusion / NPP, enhanced sampling, adaptive partitioning, and multi-time-step integrations. Each of these on their own can provide massive speed ups. In fact, the user determines the desired resolution which depends on the system and question of interest. As illustrated below, reducing the resolution and thus system complexity from all-atom to globular beads can increase the model’s speed from anywhere from one to three orders of magnitude. As the tech matures and the field standardizes, further speed gains will be made in combining multiple methods.
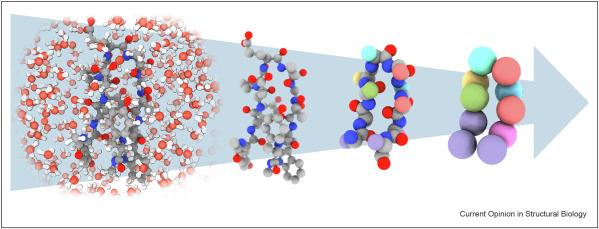
In principle, it’s relatively straightforward to go from full specificity to abstractions – and recent technical advances have made that process 10x and then 100x more data efficient. It’s intuitively more challenging to enable the “zoomed in” abstractions of a specific system to be transferable to other systems. With CG models, the units are no longer individual atoms but vague, abstracted globs. And, the further one coarse grains the more information or degrees of freedom are discarded. Groups have started to make meaningful progress towards this in the last two years by representing the effective CG energy by a “deep GNN, capturing multi-body terms without imposing restrictive low-dimensional functional forms”. They developed a transferable coarse grained (CG) model at the resolution of full backbone heavy atoms able to predict folded structures, intermediates, metastable and unfolded basis, and fluctuations of intrinsically disordered proteins for proteins outside the training set with low sequence similarity (<50%). They even hint that a “universal” CG model is possible.
An analogous area of cutting edge research is seamlessly transitioning between levels of abstraction. Researchers are working on enabling NNPs and FFs to talk the same language to better communicate with one another (FFs are expressed in terms of charges, bonds, electrostatics, etc., whereas NNPs only know coordinates and energies). Others recently figured out how to go backwards from a CG model to an all-atom one. Still others dream of recursively coarse graining to arbitrarily large macrostructures or time scales, “perhaps one day encompassing complexes of multiple proteins, cell membranes.”
Conclusion
At Compound, we spend an immense amount of time trying to understand major technical inflection points that one can build applied research companies around. We look to invest around those inflections when a hard technical problem can be de-risked and a product can be iterated upon in 2-4 years.
NNPs feel primed for this as deep learning and ground truth physics are increasingly converging, and that gap will be closed primarily by better physics modeling more so than pure AI. In such a world, the computational stack will be AI for high throughput ideation and NNPs/MD for experiments.
The models are at the perfect place on the technological S-curve to form a startup around: they’re showing glimpses of already being useful for some small subset of applications and appear several years away from being truly and broadly practical. They may prove to be similarly as important as AI models more broadly, but have garnered a tiny sliver of the hype.
Appendix
Other miscellaneous open questions not mentioned throughput this piece include:
- How can we check ground truth accuracy of NNPs without going back to generating DFT data as that defeats the purpose?
- Improvements in metadynamics, enhanced sampling
- How much of remaining accuracy issues are errors that are relatively easily fixed like the systemic bias paper?
- Will generative capabilities be possible? Does it even make any sense to use NNPs / physics modeling for such a purpose or should you always return to AI models for that task? Maybe automated optimization of AI generated structures?
- Will physical experiments eat away at too much of their use case in bio?
- Will automated parameterization / refitting of traditional force fields progress surprisingly fast so as to lessen the relative usefulness of NNPs?
- How much of wet lab experimentation can be replaced by the computational stack above within the next 10 years? Within 20?
- Will it be possible to train a model on a given subset of elements and then change that subset without having to retrain the model from scratch?
Technical Details and Relative Performance of Top Models